


据储能界了解到,
本文亮点:深度势能模型(Deep Potential, DP)通过先进的机器学习技术,从大量的原子结构和能量数据中提取知识,构建出高精度的势能面。这一创新方法有效突破了传统力场方法的局限,为材料科学领域带来了新的视角。本文概述了DP模型的基本原理,并回顾了其在电化学储能材料中的应用,包括负极材料、正极材料、固态电解质和电解液。展示了其在揭示电池材料微观结构和动力学行为方面的优势。同时,指出了DP模型在电池材料模拟中仍需改进的问题,并展望了其在电池材料设计和优化中的潜在应用前景。
摘 要 深度势能模型(deep potential,DP)通过先进的机器学习技术,从大量的原子结构和能量数据中提取知识,构建出高精度的势能面。这一创新方法有效突破了传统力场方法的局限,为材料科学领域带来了新的视角。本文概述了DP模型和软件的基本原理、开发进展与应用流程,回顾了其在电化学储能材料设计中的应用,展示了DP模型在揭示电池材料微观结构和动力学行为方面的优势。在正负极材料的研究中,精确描述脱嵌锂过程中材料的结构演变和自由能变化;在固态电解质的研究中,精确描述了材料结构与离子输运行为;在电解液的研究中,不仅提高了对溶液动态结构和性质的认识,还为氧化还原电位、酸度等物理化学性质的精确计算提供了新策略;在界面的研究中,准确解析了界面形成过程中的结构演变以及性质。这些对材料的准确描述均有利于加速对能源材料的开发。同时,指出了DP模型在电池材料模拟中仍需改进的问题,并展望了其在电池材料设计和优化中的潜在应用前景。结果说明,深度势能模型作为一种强大的计算工具,在电化学储能材料的研究中展现出巨大的应用潜力。通过不断的模型优化和算法创新,DP模型有望在未来的材料设计和电池技术发展中发挥更加关键的作用。
关键词 深度势能;分子模拟;储能材料;神经网络
在材料科学领域,分子模拟技术正逐渐成为研究和开发新材料的重要工具。分子模拟的核心挑战在于准确且高效地描述原子间的相互作用,这通常通过求解势能面来实现。传统的量子力学方法,如密度泛函理论(density functional theory,DFT),虽然在预测材料性质方面极为精确,但其计算成本随着系统规模的增加而急剧上升,限制了其在大规模和长时间模拟中的应用;基于经验参数的力场方法虽然效率足够,但精度不足。为了解决这一问题,研究者们一直在探索能够平衡计算精度和效率的新方法。近年来,随着机器学习技术的飞速发展,深度学习在多个领域展现出了巨大的潜力,特别是在图像识别、自然语言处理等方面取得了显著的成就。受此启发,研究者们开始尝试将深度学习技术应用于分子模拟中,以期开发出既准确又高效的新型势能面模型,即深度势能(deep potential,DP)模型。与传统的经验势函数相比,深度势能模型能够通过学习大量的原子结构和相应的能量、受力数据,构建出更为精确的势能面,利用深度学习的强大非线性拟合能力,直接从数据中学习原子间复杂的相互作用。深度势能模型的发展可以追溯到早期的神经网络势函数(neural network potential,NNP),如Behler和Parrinello提出的BP-NNP。这些早期模型通过将原子的局部环境信息编码为神经网络的输入,成功预测了材料的力学和热力学性质。随着深度学习技术的进步,更多的深度势能模型被开发出来,如Schtt等提出的SchNet等,这些模型通过更为复杂的神经网络结构,进一步提高了预测的准确性。
DPA-1和DPA-2是深度势能模型在大原子模型层面的两个重要里程碑。DPA-1通过引入注意力机制,增强了模型对原子间相互作用的描述能力,实现了对元素周期表上大多数原子的覆盖。DPA-2则进一步扩展了模型的适用范围,通过大型原子模型架构,进一步处理包含多种元素的复杂材料体系。不仅提升了深度势能模型的精度,还大大降低了训练过程的花费。深度势能模型的发展不仅在理论上取得了突破,而且在实际应用中也展现出了巨大的潜力。通过将物理建模、机器学习和高性能计算技术相结合,深度势能模型已经在最先进的超级计算机上成功应用于超过1亿原子的分子动力学模拟。这种技术的进步,正在推动材料科学进入一个新的研究阶段,为材料设计和发现提供了新的工具和方法。
在电化学储能领域,正极、负极以及电解质等关键材料的性质,对电池的整体表现起着决定性作用。尽管基于密度泛函理论(DFT)、经典分子动力学(CMD)和从头算动力学(AIMD)的传统计算模拟方法已广泛用于这些材料的研究,但它们难以在确保模拟精度的同时,覆盖足够长的时间尺度,限制了对材料性质的深入理解和预测。引入深度势能模拟,能够有效克服传统方法的局限,不仅提供了对电池材料性质更深层次的洞察,而且有力推动了计算模拟在电池材料研发中的应用。机器学习算法已在新能源领域获得较多应用,随着DP技术的不断发展和完善,预计其将在电池材料的设计和优化中扮演越来越重要的角色。通过更深入地理解材料在原子尺度下的行为,DP技术将有助于揭示电池充放电过程中的复杂机制,为开发具有更高能量密度、更长循环寿命和更安全性能的电池材料提供理论支持和指导。因此,DP技术的应用前景广阔,将为电池材料的创新研发开辟新的道路。本文主要介绍了深度势能模型在电池材料领域的应用,并阐述了其问题和发展方向。
1 深度势能简介
分子动力学模拟需要用力场模型确定原子的能量和受力,进而模拟原子分子体系随时间的动态变化。构建力场有两种思路,一是基于量子化学方法获得当前体系结构下准确的势能和势能梯度(受力),并将其作为力场模型使用,该方法被称为第一性原理分子动力学(ab initio molecular dynamics,AIMD)。该方法足够准确,但计算量非常高,其时间复杂度在O(N3)以上。巨大的计算开销使得AIMD的典型应用局限在了对几百个原子进行数十皮秒的模拟。另一种思路是使用一种近似的势函数,并使用实验或精确计算的结果来调节力场参数。这样得到的力场称为经验力场。一种经典对相互作用力场的函数形式见式(1):
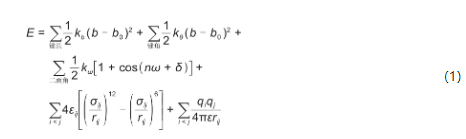
分别用弹簧势描述分子内键长,键角,二面角等相互作用,用Lennard-Jones势描述原子间范德华相互作用,用点电荷库仑势描述原子间静电相互作用。简单的函数形式使得经验力场的计算效率非常高,其时间复杂度一般不超过O(N2)。但经验力场只是一个粗糙的物理模型,为了计算速度牺牲了一定的可靠性,不能胜任某些需要精确估计能量的场合。
因此,开发兼具精度和效率的力场具有重大意义。经验力场虽然精度不足,但其采用原子位置函数来近似表示真实势能面的思路仍具有很大潜力。真实的力场可看作一个非常复杂的高维函数,经验力场的函数形式过于简单而引入了很大误差,但若使用包含更多参数的更复杂函数形式,并用机器学习方法直接模拟真实的势能面,则有望在避免量子化学层层迭代的昂贵计算的同时,相比经验力场更好地拟合真实势能面。经过实践,用深度神经网络模型学习得到的深度势能力场是应对该问题的有效解决方案。下文将对其进行简要介绍。
1.1 深度势能基本理论
深度势能理论是一种利用深度学习方法来准确预测材料体系中原子受力和能量的方法。该方法的核心是构建神经网络实现原子能量、受力和原子局部环境的映射。使用深度神经网络的方式、通过机器学习拟合势能面得到的力场模型,能够在保持计算效率的同时逼近量子化学计算精度。
1.1.1 深度神经网络(DNN)
(1)可扩展性
如仅采用如图1所示的简单神经网络模型,输入变量的维度即不可改变,即用N个原子训练的模型只能应用于N个原子的体系,若要其能用于其他原子数(如N+1),则需要两个方面的改进,一方面需要神经网络结构相比多层感知机更加灵活,另一方面需要将能量分解到每个原子上,将每个原子作为单元的能量贡献,再将其求和。
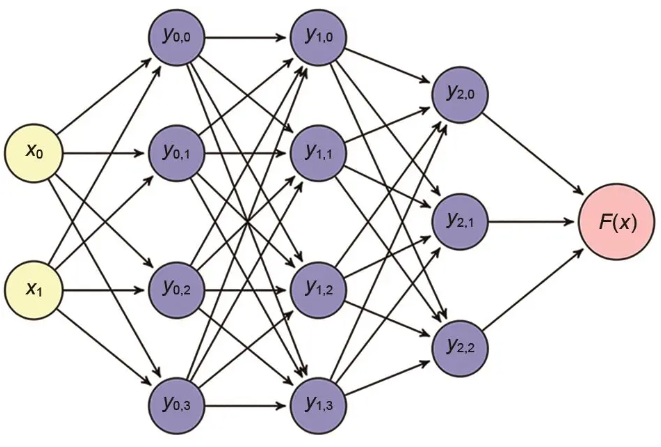
图1 多层神经网络
(2)平移和旋转对称性
分子动力学模拟中,体系性质只与盒子中原子间的相对位置有关,原子和坐标轴的设置不影响对体系结构的描述。另外,体系整体的平移和旋转也不对体系性质造成任何影响。即对于一组原子坐标(r1⋯rN),经过平移、旋转操作变换为(r′1⋯r′N)时,应有(r1⋯rN)=(r′1⋯r′N)。
(3)交换对称性
分子动力学模拟中,相同的原子交换位置时,不对体系造成任何影响。如一个水分子坐标为(r1, r2, r3),其中r1为氧原子坐标,r2和r3为氢原子坐标,(r1, r2, r3)和(r1, r3, r2)描绘的是相同的分子。因此,深度势能模型既要区分不同原子类型,还要满足相同原子交换不变性。
在满足以上要求后,构建出数学形式如式(2)的深度学习势函数。

首先,根据可拓展性的要求,体系总能量为各原子能量贡献E(i)的加和,而每个原子能量贡献与其周围一定距离内的其他原子强相关,因此不仅需要原子本身的位置信息,还需要输入原子周围环境的信息Dαi(r)。此处引入了一个假设,即原子能量贡献只与离该原子较近的其他原子有关,而与距离更远的原子无关,因此指定一个临界距离rc,对于原子i,其邻居原子定义为n(i)={J:rijrc}。能量项Nαi[Dαi(r)]输入为ri,{rj:J∈n(i)}。该模型中忽略了距离较远原子之间的影响,这一假设已在金属、固态电解质、电解液体系中被证明是一个合理的近似。
1.1.2 外层神经网络
Nαi()是一套神经网络,用于输入预处理后的原子坐标并输出能量。对于每个中心原子使用相同的网络构架,对于不同的中心原子类型使用不同的参数,从而能够区分不同的原子。这样的结构在保证了交换对称性的同时保障了模型的分辨能力(输入不等价坐标时能输出不同能量)。
1.1.3 描述子
描述子Dαi(r)起到的作用是对原子坐标进行预处理,使其满足平移、旋转对称性。其包含以下功能和特点:
(1)确保平移、旋转对称性,即坐标r经历平移、旋转变化变为r′后,有Dαi(r)=Dαi(r′)。
(2)确保模型的可拓展性,输入任意大小坐标r后,输出尺寸保持统一。
(3)保证模型的分辨能力,与外层神经网络类似,描述子内部也有一套神经网络,所有网络结构相同并固定,但不同的原子类型αi具有不同的网络参数。
1.1.4 大原子模型
由于以上介绍的DP模型适用化学范围较小,面对一个新的复杂体系时,研究者需要获取大量数据从头训练一个深度势能模型。随着大量已标记数据的积累,构建“通用”的深度势能模型,并通过预训练+微调的策略训练适合具体体系的模型能大大提高效率。DPA-1模型引入了注意力机制,根据原子间距离和角度信息重新加权得到原子间相互作用,模型能根据原子局部结构和原子间相互作用调整其表示,从而更准确地表示系统的特征。同时发现覆盖大量元素类型的大模型中,模型元素信息在可视化后,在空间呈螺旋状分布,与元素周期表位置一一对应,表明预训练模型具有良好的可解释性。
在此基础上,为进一步实现大原子模型目标,开发了DPA-2模型,其采用更为复杂的模型构架,包含单原子通道、双原子通道和螺旋双原子通道,三个通道具有不同的作用,协同提高了DPA-2模型的预测能力。单原子通道编码原子的物理特性,捕捉单个原子的特征;双原子通道含原子之间的距离、角度和键长等关系,用于捕捉原子间相互作用;旋转双原子通道在此基础上加入了旋转等变形,进一步提高了对晶体结构和对称性分子的描述能力。因此,DPA-2能够更全面地捕捉体系的化学信息,从而大大提高了模型的表示能力。
普通DP模型,DPA-1和DPA-2随样本数增加的模型精度变化趋势如图2所示(1 meV/=10 meV/nm),可见预训练模型+微调的策略大大提升了训练效率,节约了标记样本的花费。
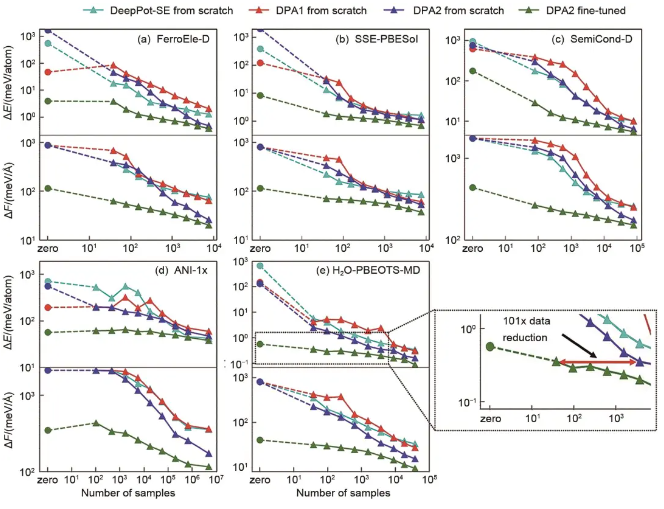
图2 下游任务样本效率对比分析。横坐标表示所需的下游数据量,纵坐标表示RMSE能量或力预测的收敛性
1.2 深度势能的开发与应用
深度势能模型的开发和应用是实现材料设计和化学过程模拟自动化的关键步骤。这些模型通过学习大量的化学数据,能够预测分子和材料的物理化学性质。
1.2.1 数据集构建
DP数据集的构建分为两个部分:①提供原子结构信息;②提供结构对应的能量、原子受力和维里张量等信息。获取后者的方法也称为标记。实际计算中通常使用DFT方法计算体系能量、原子受力和维里张量进行标记。常用软件包括VASP、CP2K、Gaussian、QE和ABACUS等。DFT数据精度决定了DP模型精度的上限。提高DFT精度一方面可使用精度更高的理论方法如杂化泛函甚至Post-HF方法;另一方面要使用更加完备的基组和更高的K点密度。但更高的DFT计算等级也带来了更多的计算开销,需要在精度和效率之间取舍。
1.2.2 模型训练
模型训练使用DeePMD-kit软件包,训练过程涉及能量、原子受力和维里张量等标签,其中最重要的是原子受力标签,一方面相比能量能提供更多信息(包含每个原子三个方向的力),另一方面梯度信息有助于避免训练时的过拟合。DeePMD-kit中需要选择描述子和训练步数,共同决定了深度势能模型的质量。DeePMD-kit中还提供了许多可调超参数,包括神经网络结构、损失函数因子等。一般深度势能模型质量对这些超参数不十分敏感,默认设置能满足一般训练的要求。
1.2.3 模型评估
模型训练后需要对获得模型进行评估以验证其精度。有两种基本方法:
(1)构建一个不包含在训练集和验证集中的测试集,在该测试集上使用dp test功能计算深度势能模型和DFT的偏差,一般能量和受力偏差分别小于10 meV/atom和100 meV/时,说明DP预测结果良好。
(2)通过深度势能分子动力学模拟得到一些物理性质与实验或DFT结果对比,包括径向分布函数、密度、扩散系数、电导率和黏度等。
1.2.4 模型推理
在得到势函数模型后,用此模型计算输入的体系结构信息而得到能量、受力、维里张量等输出信息的过程称为模型推理,DeePMD-kit软件提供了Python和C++等多种语言接口,与分子动力学模拟软件联用即可将模型推理获的受力等信息用于分子动力学模拟中运动方程的计算。目前支持的软件则主要有Lammps、ASE、i-PI和Gromacs等。
1.3 深度势能相关软件与平台
深度势能相关软件与平台的发展,为化学和材料科学的研究者提供了前所未有的计算能力和灵活性。这些工具不仅加速了科学研究的进程,还拓宽了研究的可能性。
1.3.1 dpdata
dpdata是一个用于结构和标记数据格式转化的开源免费程序包,用Python语言写成,可将VASP、Gaussian、CP2K、QE、ABACUS、SIESTA、FHI-aims、PWmat等第一性原理程序的计算结果文件转化为DeePMD-kit可识别的格式,作为标注训练集。也可进行扩胞、移动原子、更改原子等结构操作和结构文件在不同软件间转化。
1.3.2 DeepMD-kit
DeepMD-kit是一个构建原子分子体系深度学习神经网络势函数并进行分子动力学模拟的开源免费程序包。DeePMD-kit提供了一套完整的工具集,与TensorFlow接口或PyTorch接口结合实现了高速模型训练和推理,训练过程可通过DP-GEN实现自动化。模型推理使用C++或Python接口,输入原子位置信息,即可返回当前结构下的能量和原子受力,使用C、C++或Python语言编写的MD包可与DeePMD-kit联用进行分子动力学运动方程求解。与LAMMPS软件包联用时可实现GPU加速,因此这一组合应用最为广泛。目前,DeepMD-kit推出了3.0版本,增加了对DPA-2大原子模型的支持。
1.3.3 DP-GEN
DP-GEN是一个实现深度势能并发学习的开源免费程序包,由Python语言编写,能在超算集群上自动准备任务脚本并进行分布式计算,适用Slurm、PBS、LSF等多种任务队列,可实现深度势能模型的自动化采样与训练,自动化流程如图3所示,分为初始训练集构建、训练、势能面探索、标记几个步骤,以下将逐一介绍。
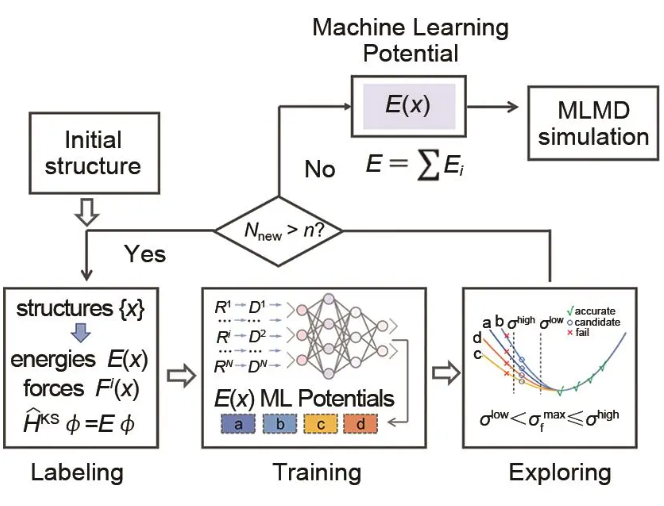
图3 DP-GEN自动化流程
(1)初始训练集构建
并发学习训练流程中的首轮训练需用用户自己提供初始数据集,虽然最后收敛的模型结果对初始训练集并不敏感,但合理构建的初始训练集能有效提高训练效率。初始训练集一般需要含有几百个训练数据,并要尽可能充分覆盖所关心的势能面区域。初始训练集一般可采用AIMD生成。相比于固态材料体系,电解液体系构象空间更加复杂,对于初始训练集的要求也更高。可使用AIMD结合CMD,配合适当的采样间隔获取初始结构,且需要包含不同浓度下的数据以充分采集离子靠近或远离时的结构。
(2)标记
初始结构或每轮被挑选出的结构需做DFT做单点能计算以得到其对应的能量和原子受力。标记任务中除设置合适的DFT理论等级外,还需要设置每轮标记的数目,数目过多则采集过多重复结构而浪费标记算力,采集过少则新增样本不足而需要更多轮迭代,浪费探索和训练算力。标记数目一般设置为100个。
(3)训练
训练过程中调用DeepMD-kit对初始数据集(第一轮训练)或上一轮迭代后获得的所有数据集进行训练,通过将能量和原子受力的损失函数最小化来构建体系结构到能量和原子受力映射。用户需要选择合适的深度势能模型和描述子,描述子截断半径一般设置为0.6 nm。训练步数一般为105~106量级,训练过程中可使用较少步数,待DP-GEN收敛后再进行一轮106步的长训以增加模型精度。为了在DP-GEN并发学习中获知模型对势能面的描述情况,共训练了4个模型,4个模型使用的训练集相同,仅神经网络中使用的激活函数不同,通过4个模型预测的原子受力的最大均方误差σfmax衡量模型预测效果。
(4)势能面探索
势能面探索过程使用上一步训练好的深度势能模型,对指定初始结构进行分子动力学模拟,使用4个模型预测的原子受力的最大均方误差σfmax衡量模型预测效果,用户置信区间的上下限分别被定义为σhigh和σlow,如某一结构的σfmaxσlow则表明模型已充分学习了该结构;如σfmaxσlow表明结构已被模型学习到;若σfmaxσhigh则表明结构超出已学习势能面过多,结构可能不合理结构而影响DFT收敛;如σfmax在σhigh和σlow之间则表明选定结构略微超出已学习势能面,是应当标记的结构。势能面探索过程应当使用多个温度、压力、物质组成(如浓度、配比等)条件,以充分学习模型需要覆盖的势能面区域。
1.4 OpenLAM
OpenLAM为“大原子模型计划”,口号是“征服元素周期表”,旨在通过建立开源开放的围绕微尺度大模型的生态,为微观科学研究提供新的基础设施,并推动材料、能源、生物制药等领域微尺度工业设计的变革。最新发布的深度势能预训练大模型DPA-2已成为大原子模型的重要载体,其在模型架构显著更新的同时,最大的特点在于采用了多任务训练的策略,从而可以同时学习计算设置不同、标签类型不同的各类数据集。由此产生的模型在下游任务上显示出极强的few-shot乃至zero-shot迁移的能力,显著超越过去的方案。目前用于训练DPA-2模型的数据集已覆盖了半导体、钙钛矿、合金、表面催化、正极材料、固态电解质、有机分子等多类体系。
1.5 AIS square
AIS square中文名称为科学智能广场,旨在打破传统的“作坊式”和“发散式”科研模式,提供一个跨学科的科研平台。用户可以在平台上上传和贡献科学计算数据集、模型及工作流,同时也可以一键下载和使用已经训练好的专用模型。此外,平台还支持应用预训练大模型,通过少量有针对性的数据进行微调,从而方便、快捷地获得下游任务所需的模型,降低计算代价和研究成本。目前,科学智能广场已经获取并向公众共享了总机时价值超3亿核时、覆盖各个应用领域的模拟仿真数据,并贡献了50多个特定场景专用模型。这些模型涵盖了从材料结构、力学、热力学性质计算到发动机喷雾燃烧过程模拟等广泛的科学计算领域。
1.6 模型蒸馏
预训练模型为了追求模型泛化能力,模型参数规模较大,从而影响了模型推理的计算速度。为了提高其在真实场景中的计算速度,使用了模型蒸馏的方法。模型经预训练和少量下游体系数据微调后得到Teacher模型(如DPA-2)。之后使用类似主动学习的策略,用Teacher模型探索势能面空间并标注(替代直接做DFT),从而获得一个更加简单和轻量的Student模型(如DPA-1、DeepPot-SE等),蒸馏后的Student模型在精度接近Teacher模型的基础上,效率提升了两个数量级。实现了知识从高泛化能力的复杂模型到简单高效简化模型的知识蒸馏。
2 深度势能在电化学储能材料中的应用
2.1 负极材料
锂离子电池的成功商业化,起始于石油焦负极材料。负极作为锂离子电池必不可少的关键材料,目前主要集中在碳基、金属锂和硅基等合金类负极,采用传统的石墨负极可以基本满足消费电子、动力电池、储能电池的要求,采用合金类负极材料有望进一步提高能量密度,全固态电池的研发则有望推动金属锂负极的应用。此外,在钠离子电池中,无序度较大的无定形碳基负极材料具有较高的储钠比容量、较低的储钠电位和优异的循环稳定性,是最有应用前景的钠离子电池负极材料。目前,深度势能分子动力学模拟方法已被应用于研究锂金属、硅负极、碳负极材料储锂或储钠过程的结构演变和扩散动力学等行为。由于这些负极材料主要是金属或无机晶体材料,其DP开发可以很自然地借鉴过去金属和合金领域DP开发的思路。
2.1.1 碳基负极材料
碳基材料在二次电池中应用非常广泛,根据碳原子中电子之间不同的轨道杂化形式,可以将碳材料主要分为sp、sp2和sp3碳材料。碳基负极材料以sp2杂化为主,包括石墨、无定形碳和纳米碳材料。由于结晶度和碳层排列方式的不同,它们的物理性质、化学性质和电化学性质等都呈现不同的特点。王松有团队开发了一个碳基材料的DP模型,训练范围覆盖碳的晶相和液相,其中晶相类结构包括12种不同的体相或低维碳结构(例如石墨、金刚石、碳纳米管、无定形碳),液相类结构则考虑了4种不同密度的液态碳结构(1.7 g/cm3、1.9 g/cm3、2.6 g/cm3、3.2 g/cm3),总计约29000个数据点。他们使用DP模型研究了不同结构的碳材料的状态方程,单层石墨烯中各类缺陷的形成能[Stone-Wales (SW) defect,单空位、双空位、三空位和四空位]、不同密度的液态碳和非晶碳的结构。结果表明,DP模型能够高精度地描述绝大多数碳材料的状态方程(除了sc、fcc、bcc等几类高能量不稳定的碳构型)、单层石墨烯中缺陷结构的热力学稳定性,以及液态碳和非晶碳的结构和成键特征,并且在一些未纳入训练集中的碳结构也具有较好的迁移能力。同时,作者也指出DP模型目前还存在一个问题,即无法准确描述石墨烯层间的范德华作用。孙强团队结合密度泛函理论计算和CALPSO结构搜索方法设计了一类热力学和动力学稳定的3维多孔的硫化碳材料C8S,因其具备高的克容量、低的迁移势垒、低的嵌钠电压和小的体积变化,可用于钠离子电池的负极材料。为研究Na+在C8S中的扩散性质,他们开发了一个训练数据集中包含20000个结构的DP模型。如图4所示,结果表明,当钠离子浓度较低时,钠离子的扩散系数为3.2310-7 cm2/s,而当钠离子浓度较高时,钠离子的扩散系数降低为2.3610-8 cm2/s,两者相差一个数量级,这与高浓度时Na+之间的强的排斥作用和Na+穿过六边形石墨环时较高的迁移势垒有关。
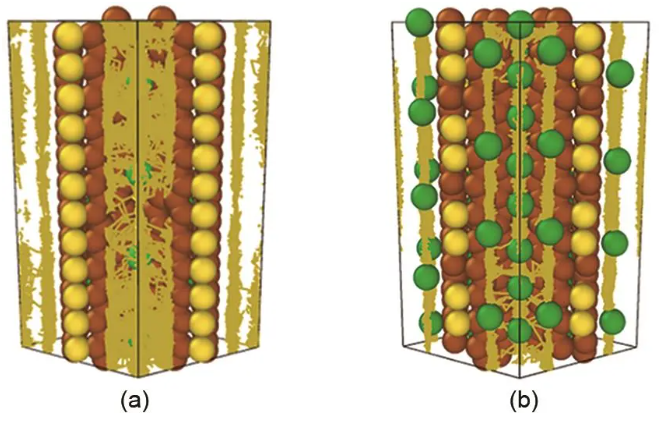
图4 (a) 低Na和 (b) 高Na含量C8S材料中Na+的扩散轨迹
2.1.2 硅基负极材料
硅由于其极高的理论容量被认为是下一代高比能锂电池最理想的负极材料之一。然而,硅负极充放电过程中存在巨大的体积膨胀和结构变化,活性材料颗粒容易粉化和失去电接触,最终导致容量快速衰减,严重阻碍了Si基负极的实际应用。为了更好地开发Si基负极,有必要了解其锂化/脱锂过程的反应机制、微观结构的演变规律和锂的扩散动力学行为。何奕团队开发了一个Li-Si二元体系的DP模型,训练范围涵盖了从Li/Si比例为0~4.2的9种Li-Si体系(Si、LiSi3、LiSi、Li13Si4、Li7Si2、Li21Si5、Li、LiSi64和Li54Si),并且同时包含晶相、液相和非晶相等丰富的结构,通过使用主动学习策略来进一步提高数据集的质量和DP的预测能力。他们使用DP进行了分子动力学(MD)模拟,以研究Li在非晶态Li-Si系统中的扩散性。结果表明,DP能够以接近量子力学计算的精度预测Li-Si体系的体积变化、径向分布函数和Li在非晶态Li-Si中的扩散性,同时模拟速度比从头算分子动力学模拟快20倍。傅方佳等开发了一个高精度的Li-Si体系的DP模型,训练范围扩大到Li/Si比例为0~4.5的15种Li-Si体系(bcc, fcc 和 hcp Li;Fd3̅m Si of diamond 和 I41/amd Si;LiSi3, LiSi, Li12Si7, Li2Si, Li7Si3, Li13Si4, Li7Si2, Li15Si4, Li21Si5, 和 Li22Si5),并且同时包含晶相、液相和非晶相等丰富的结构。如图5所示,他们结合DeePMD(深度势能分子动力学)和GCMC(grand canonical Monte Carlo,巨正则蒙特卡洛)方法,模拟了晶体硅(c-Si)和非晶硅(a-Si)在锂离子电池充放电过程中的嵌锂/脱锂电位、微观结构演化和应力分布。研究发现,c-Si和a-Si锂化之间平台高度的差异机理是因为c-Si中初始Li嵌入的能量低于a-Si中初始Li嵌入的能量,而c-Si和a-Si都锂化反应到相似的a-LixSi相,有限电压滞后的机制来源于c-Li3.75Si到a-Li3.75Si相转变相关的潜热效应,a-Si嵌锂时产生的应力要比c-Si更低,且锂的扩散有利于减少应力。
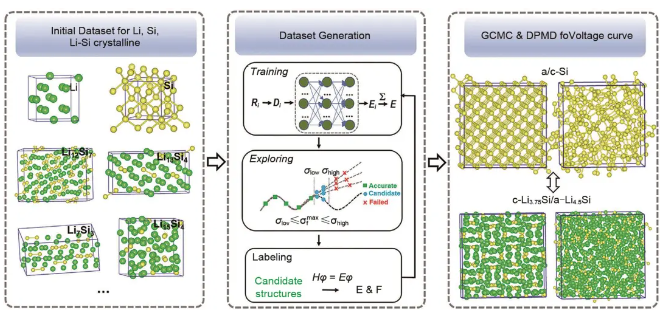
图5 Li-S-Si体系DP模型训练及检验过程
2.1.3 锂金属负极
锂金属电池是实现高能量密度电池的理想路径,然而锂枝晶的生长及其带来的安全问题限制了锂金属电池的实际应用,理解锂原子沉积过程枝晶的生长机制有助于设计策略抑制枝晶生长。郑家新团队开发了一个锂金属表面的DP模型,训练数据集包括近10000种体相结构(bcc Li)和6000种表面结构,包含bcc Li的三种低指数晶面[100]、[110]和[111]。他们构建了超过10万原子数的锂金属表面结构模型,并使用DPMD进行了长达3 ns分子动力学模拟来模拟锂原子在锂金属表面的沉积过程。如图6所示,研究发现,锂金属在沉积过程中存在两种自愈合机制,第一种是表面愈合机制,指在锂原子均匀沉积的情况下,锂金属表面的缺陷(如凹坑或不规则性)会自动被填充,形成平滑表面的现象。第二种是体相愈合机制,指在锂金属的非均匀沉积过程中,由于电流密度不均匀导致的锂枝晶生长,当两个枝晶相遇时,它们会融合并愈合成一个更大的枝晶,同时消除了它们之间的空隙。考虑到锂金属的模量、弹性各向异性和表面扩散等性质对于抑制枝晶生长非常关键,而已有DP模型在应力和弹性常数的预测上仍然不够准确,Viswanathan团队独立开发了一个高精度的锂金属DP模型,训练集覆盖了体相(bcc Li,fcc Li,hcp Li)、熔融液相、表面bcc [110],[100],[111],[210],[211],[221]和缺陷(间隙、空位和晶界)等多类结构,总计5053个构型。他们使用DP模型准确预测了锂金属的热力学性质、空位形成能、状态方程、声子谱、有限温度下的弹性常数、表面能和沃尔夫构造。结果表明,相较于低指数晶面,高指数晶面由于配位数少导致锂原子吸附能较大,且通常具有较大的锂原子表面扩散能垒。
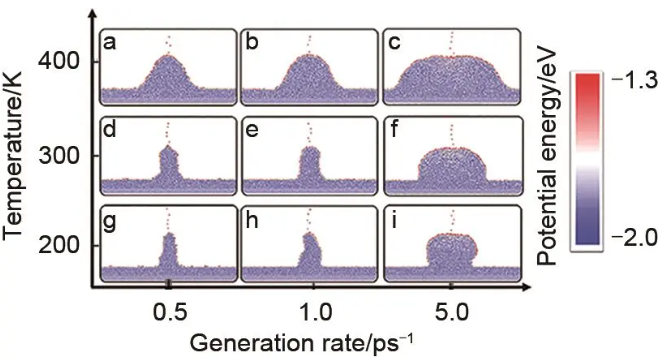
图6 不同温度(T)和生成速率(Rg)下不均匀沉积快照。Rg=0.5 ps-1 (a, d, g),1.0 ps-1 (b, e, h) 和5.0 ps-1 (c, f, i);T=400 K (a~c), 300 K (d~f), 200 K, (g~i)
2.1.4 无负极体系
铜是锂离子电池行业中最常用的集流体材料,已有多项研究报道了使用多孔铜集流体来容纳锂的沉积并抑制枝晶生长。为理解Li在Cu表面的沉积行为,郑家新团队开发了一个Li-Cu二元体系的DP模型,训练范围覆盖金属铜、金属锂、四种Li-Cu二元金属间化合物(I4/mmm-Li3Cu,I4/mmm-LiCu3,Pm3m-LiCu3,Fm3m-LiCu3)和四种Li/Cu界面结构Cu [100]-Li, Cu [110]-Li, Cu [111]-Li, 和Cu [211]-Li。他们使用DP模型准确预测了Li-Cu二元金属间化合物的平衡体积、状态方程和弹性常数,并研究了Li原子在金属Cu表面的吸附能。这些结果证实了DP模型可用于模拟锂原子在金属Cu表面的吸附、扩散动力学行为,将为研究锂金属电池中Li-Cu界面问题提供一个有效的工具。
2.2 正极材料
正极材料是锂离子电池中的重要组成部分,它们直接影响电池的能量密度、循环稳定性、成本和安全性,通过提高使用电压或者寻找新的高能量密度正极材料一直是学术界和产业界的重心。目前,锂离子电池中商用的正极材料有LiCoO2、LiFePO4和Li(NixCoyMn1-x-y)O2;钠离子电池正极目前还未完全实现商用化,研究中关注的正极材料主要包括氧化物类、聚阴离子类、普鲁士蓝类等。由于正极材料一般都含有过渡金属,往往采取DFT+U的方法来研究,在构建DP训练集时可能存在一定困难,导致DP在正极材料中的应用还较少。
2.2.1 锂离子电池正极材料
高镍正极材料在高荷电状态下会发生结构相变和体积坍缩,严重影响电池性能。为理解体积坍缩和结构相变之间的关系,Hakim Iddir团队构造了LiNiO2材料处于高荷电状态下的两类分相结构模型(富O3和富O1),并分别训练了这两类结构在多个浓度下(LixNiO2,x=0, 0.04, 0.08, 0.12, 0.17)的DP模型。他们使用DP模型做长时间的分子动力学模拟,获得了两类分相结构模型在不同组成范围(LixNiO2,x=0, 0.04, 0.08, 0.12, 0.17)和温度范围下(298.15~350 K)的焓和Gibbs自由能,通过比较发现两类结构模型室温下的自由能差别非常小可以忽略,由此推测实验中观察到的NiO2层堆积方式的转变(由O3到O1)可能来自动力学上的原因。
LiCoO2正极材料在高电压下工作容易发生析氧和过渡金属溶出,显著加速电池老化。为理解过渡金属溶出和析氧之间的动态关联,许审镇团队开发了LiCoO2体系的DP模型,训练数据集覆盖O1、O3和Spinel等三种结构的LiCoO2,并且考虑了锂浓度从0到1的变化范围。对于过渡金属离子,对局域环境的描述除了原子坐标,还需要考虑自旋状态,由于DP模型并没有显性地考虑自旋这一自由度,需要确保数据集中相同组成下的所有结构都处于相同自旋状态。然而常规的DFT+U计算可能会出现结构弛豫后收敛到自旋非基态的状态,为确保数据的有效性和一致性,他们发展了一套确定LiCoO2正极材料电子结构基态工作流。基于此工作流,成功构建了不同相下不同Li浓度的LixCoO2材料的DP模型。他们以O3-CoO2为模型,通过DeePMD模拟结合增强采样技术,获取了Co4+离子迁移和氧二聚体产生的动力学关联,发现过渡金属层内Co空位团簇的形成是氧-氧二聚体形成的先决条件。基于MD过程获得的路径演化信息,揭示了Ti掺杂离子对上述过程的影响,如图7所示。
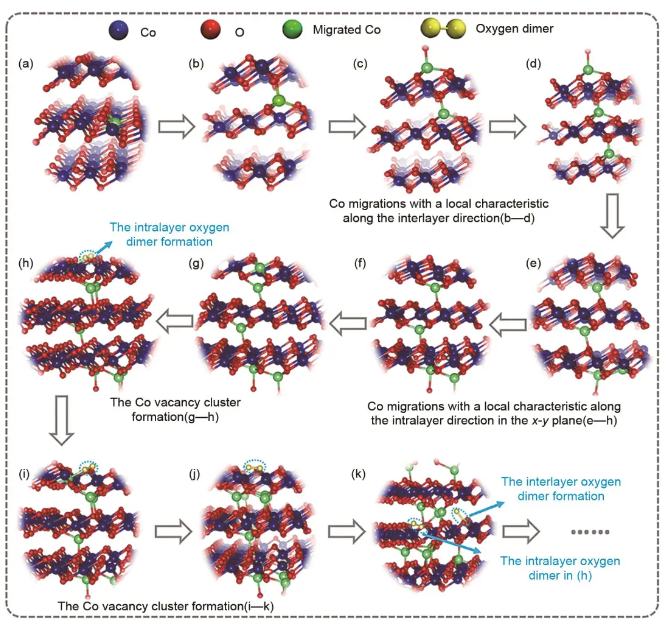
图7 基于增强采样DPMD模拟的CoO2模型产氧反应路径图
LiFePO4正极材料在不同荷电状态下实测的扩散系数差数个量级,同时相比理论计算预测的锂离子迁移势垒推导出的扩散系数低数个量级。为解释这一现象,陈胜利团队开发了一个LiFePO4材料的DP模型,并基于此计算LiFePO4中不同荷电状态下的锂离子迁移势垒。进一步,结合动力学蒙特卡洛模拟来模拟恒流充电过程中的锂离子动态分布,发现Li-Li局域配位环境(数量和构型)会显著影响锂离子的迁移势垒,并且锂离子浓度沿扩散通道的梯度分布导致锂离子沿一维通道向前跳跃和向后跳跃的迁移势垒存在非常大的不对称,导致了锂离子非常慢的扩散和扩散系数随锂浓度变化表现出数量级的显著差异。
2.2.2 钠离子电池正极材料
层状氧化物是目前钠离子电池商业化应用中非常有前景的一类层状氧化物正极材料,制备方法简单、比容量和电压较高,成本低。其中,O3相正极材料具有较高的初始Na含量,能够脱出更多的钠离子,具有较高的容量;P2相正极材料具有较大的Na层间距,能够提升钠离子的传输速率和保持层状结构的完整性,具有优异的倍率性能和循环性能。对于P2相这类高倍率正极材料,需要深入理解材料结构和钠金属离子扩散动力学之间的关系。固态核磁共振(ssNMR)技术凭借对局部环境和动态信息的独特敏感性,在电池材料的研究中得到了广泛应用。然而,因过渡金属(TM)离子与被观测核的复杂相互作用,正极材料的NMR谱峰指认往往依赖密度泛函理论(DFT)计算。对于高倍率正极材料,碱金属离子局部结构和其动态信息耦合形成的动态NMR化学位移,目前仍缺乏合适计算方法。
程俊团队发展了高倍率电池正极材料动态NMR化学位移的计算方法,应用于P2型钠离子电池正极材料,并结合ssNMR、X射线衍射(XRD)以揭示其精细结构,如TM离子超结构、TM氧化物层的堆垛以及它们和Na+扩散动力学之间的关系。如图8所示,他们首次联用DFT计算局域位点化学位移和200 ns深度势能分子动力学(DPMD)模拟得到收敛的Na+分布,计算了P2型Na2/3(Mg1/3Mn2/3)O2动态23Na化学位移。并结合ssNMR、XRD谱的堆垛精修,指认23Na NMR谱中两个化学位移峰分别来源于两种层间堆垛方式,对应空间群P63/mcm和P6322,并精确定量,修正了人们之前一直认为该类材料仅有单一堆垛(空间群P63/mcm)的认知。不过该工作中应用优化结构(0 K)来计算局域位点化学位移,忽略了有限温度下的热力学涨落对化学位移的影响。
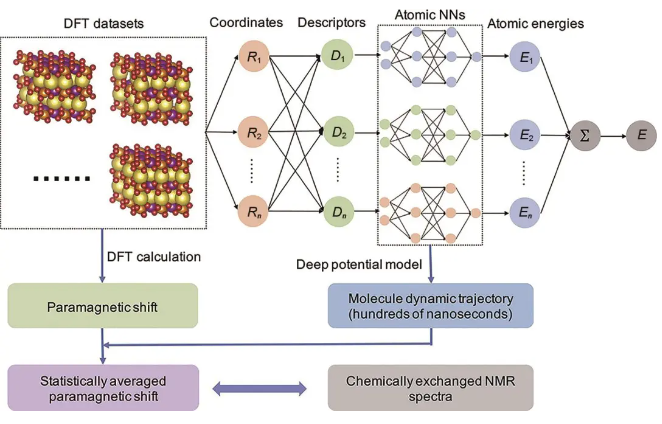
图8 基于DPMD的固态核磁化学位移计算流程
动态化学位移的直接精确计算依赖于对DPMD轨迹中大量结构做DFT计算,计算资源消耗较大,限制了方法的推广应用。为提升计算动态NMR化学位移方法的适用性,程俊团队建立了一个动态化学位移的DFT数据集,并开发了一种机器学习(ML)方法,其不仅可以进行快速构象采样,还可以高效预测化学位移,这个方法在GPU卡上计算时间与DFT方法在CPU核上计算时间相比,缩短了几个数量级。他们使用该ML方法预测了P2型Na2/3(Mg1/3Mn2/3)O2 200 ns DPMD模拟中50000个结构的23Na位移,这基本消除了动态NMR位移计算值的统计误差,且与实验测量结果一致。此外,对DPMD轨迹动态结构23Na位移做不同时间段的平均,可直接反映出动态NMR化学位移的形成过程,即Na+在不同位点之间的跳跃快慢。进一步应用该ML方法计算P2型Na2/3(Ni1/3Mn2/3)O2的动态23Na NMR化学位移,结果与实验NMR位移很好地符合,结合DFT计算证明之前指认有争议的低位移峰对应空间群P63/mcm,并揭示了不同P2型堆垛与Na+扩散系数的关系。他们发展的ML预测动态NMR化学位移方法,可用于建立高倍率电池正极材料结构、NMR谱、离子传输之间的明确关系,并可进一步推广到其他的原子核溶液体系和固态电解质中,作为联系实验NMR谱和微观动态过程的桥梁。
2.3 固态电解质
固态电解质是DPMD最早获得成功的体系之一,因其多是晶体,结构规整,且运动中主要以原子为基本单位,非常适合深度势能的原子局部近邻环境采样。深度势能分子动力学模拟已成功用于硫化物、卤化物、氧化物等固态电解质中,研究了其结构和构效关系。
2.3.1 硫化物电解质
Li10GeP2S12体系是深度势能应用较早的体系,赵金保团队、程俊团队和鄂维南团队构建了Li10GeP2S12型(包含Li10GeP2S12、Li10SiP2S12和Li10SnP2S12)电解质的深度势能模型,并对DPGEN自动采样生成势函数过程、势函数的验证和应用进行了系统性研究。在宽温度范围(300~1000 K)和大尺寸(约1000个原子)系统中,对统计误差和尺寸效应等重要技术方面进行了仔细的研究,并进行了包括不同DFT泛函、热膨胀和构型失序影响在内的基准测试。由于更充分考虑了尺寸效应和结构多样性,DPMD相比AIMD更好地重现了实验测量的扩散系数。
张强团队探索了离子在Li10GeP2S12中温度依赖的协同扩散机制。温度越高,Li扩散各向异性越弱。由于各种协同扩散模式的跳变频率对温度的线性依赖关系,扩散系数在较宽的温度范围内保持阿仑尼乌斯型温度依赖关系,加深了对温度相关离子扩散的化学起源的理解,如图9所示。David A. Egger团队更深入地研究了Li10GeP2S12的离子跳变行为,发现主晶格振动的非调和特性在实现移动离子的快速迁移中起着重要的作用。
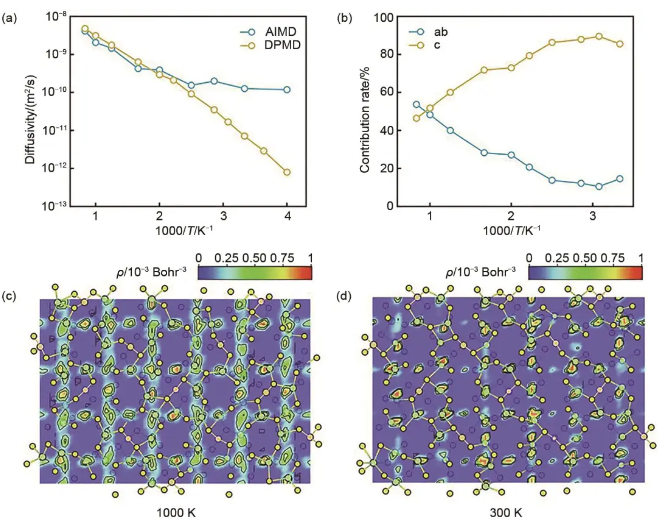
图9 Li10GeP2S12晶格中的Li+扩散行为。(a) 用AIMD和DPMD模拟的扩散系数Arrhenius图。(b) 锂离子扩散的尺寸贡献随温度倒数变化。(c) 1000 K和 (d) 300 K时Li10GeP2S12中的锂离子概率密度填色图
Li3PS4为正硫代磷酸锂体系,Michele Ceriotti团队构建了Li3PS4(包含α、β和γ相)深度势能模型,观察到的Li3PS4超离子行为的物理起源,即PS4翻转的激活驱动结构转变到高导电相,其特征是锂离子迁移位点的增加和锂离子扩散活化能的急剧降低。排除了PS4四面体在超离子相中的桨轮效应,因为PS4翻转速率和锂离子跳跃速率在熔点以下的所有温度下都有数量级的差异。文中对比了纯泛函与杂化泛函对带隙和扩散系数的模拟准确性,杂化泛函PBE0准确再现了扩散系数实验值,而纯泛函低估了带隙也高估了扩散系数,如图10所示。
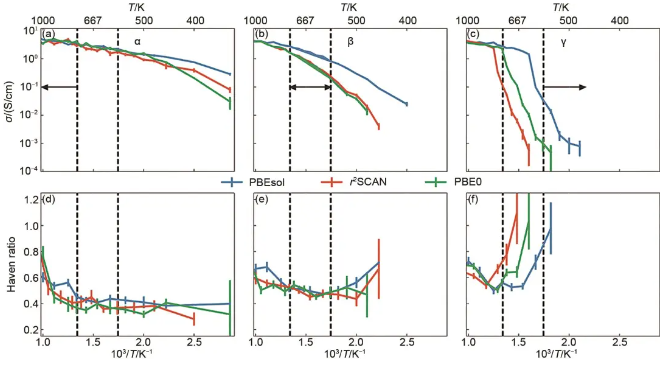
图10 电导率 (σ) 和Haven比的温度依赖性 (a, b, c) DPMD的离子电导率Arrhenius图;(d, e, f) Haven比随温度倒数变化的规律
Olivier Delaire团队采用中子散射结合DPMD的方法,研究了Na3PS4中的声子及其与Na快速扩散的耦合。发现在非调和稳定立方相的布里渊区边界处的非调和软模是控制Na3PS4中Na扩散过程的关键声子模。演示了这些强非谐波声子模式如何使Na离子沿着最小能量路径跳跃。此外,准弹性中子散射(QENS)测量,辅以从头算和机器学习分子动力学模拟,探讨了Na的扩散率和扩散机制。Li3PS4其他衍生物Na3SbS4和K3SbS4的结构和离子输运性质也已被DPMD研究,发现K3SbS4中掺杂W元素能引入空位,使K离子电导率获得一个数量级以上提升。
Li6PS5Cl及其衍生物称为锂银柱石结构,也是一类重要的固态电解质体系,Lee Sang Uck团队构建了(Li5+2x+y[A]x4+[B]y5+[C]6+1-x-yS5[D])([A] = Si, Ge, Sn; [B] = P, Sb; [C] = W, Mo; [D] = Cl, Br)电解质深度势能模型,以系统地探索混合氧化态对离子电导率的影响,从而用于高性能SSE筛选。结果显示,在混合氧化态下,SSE的电导率存在[A]4+-[C]6+[A]4+-[B]5+-[C]6+[A]4+-[B]5+[B]5+-[C]6+的顺序。[A]4+-[C]6+的组合产生了最佳性能,是由于[C]6+的固体电解质效应和[A]4+的动态晶格效应的协同作用。
Mayanak Kumar Gupta团队研究了反钙钛矿型电解质Na3FY(Y=S, Se, Te)体系中声子与扩散的关系。作为温度函数的分支分辨声子谱能量密度结果揭示了软声子模式的非调和性很大,这导致Na离子的软性动力学。三种反钙钛矿结构中,立方Na3FS体积最小,平均声子能量最大。700 K时Na3FS中的Na扩散系数约为0.63106 cm2/s,明显大于Na3FTe中的约0.16106 cm2/s,这与更软晶格导致更快扩散的假设形成鲜明对比。
2.3.2 卤化物电解质
卤化物材料由于其在离子电导率和电化学稳定性之间的良好平衡,是备受关注的固体电解质。Selva Chandrasekaran Selvaraj团队构建了Li3TiCl6的深势模型,研究在25~100 ℃ 6种不同温度下阳离子无序Li3TiCl6阴极中的锂离子传输机制,揭示了锂离子参与反相关运动,这在固态材料中很少报道。同样,锂离子动力学的自身和不同部分被用来确定Haven比率来描述锂离子在Li3TiCl6中的输运机制。从分子动力学得到的运动轨迹推断,锂离子的输运主要是通过间隙跳变进行的,这一点通过层内和层间的锂离子位移相对于模拟时间的变化得到了证实,如图11所示。
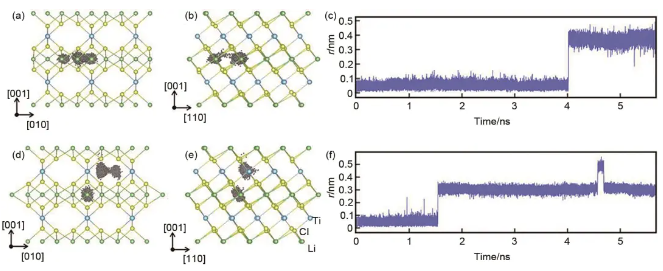
图11 沿Li3TiCl6 (a) [010] 和 (b) [110]视角方向的层内锂离子运动轨迹;(c) 对应 (a) 和 (b) 的Li原子位移随时间演化;沿Li3TiCl6 (d) [010]和 (e) [110]视角方向向的层间锂离子运动轨迹示意图;(f) 对应 (d) 和 (e) 的Li原子的位移随时间演化
Pei Yong团队研究了Li3OBr固态电解质的力学和电子特性以及Li离子的动态扩散行为,并深入探讨了缺陷浓度和分布对其的影响。结果表明,锂离子的扩散能力与缺陷浓度呈线性关系,缺陷分布对离子电导率的影响很小。在缺陷浓度为0.7%时,计算得到的Li3OBr材料的离子电导率值与之前的实验值吻合较好。(Li-Frenkel, LiCl-Schottky和Cl-O反位无序)三种缺陷中,LiCl-Schottky缺陷的存在是Li3OCl体系性能优异的主要原因,而Li空位是主要载流子。对应的钠离子电解质Na3OBr中Na离子迁移规律也被研究,计算了Na+在不同温度下的扩散系数,得到了Na+的活化能为0.42~0.43 eV。与0 K迁移势垒(0.41~0.43 eV)接近,这表明对于Na3OBr来说,有限温度效应可以忽略不计。该模型给出了外推的室温离子电导率为110-4~210-4 mS/cm,与实验结果接近。
Sun Qiang团队报道了一种新的尖晶石氯化钠(Na2Y2/3Cl4),并对其作为固体电解质的潜力进行了系统研究。尖晶石Na2Y2/3Cl4在室温下具有0.94 mS/cm的高离子电导率,并具有由面共享的八面体和四面体组成的三维各向同性扩散网络。进一步的扩散机制分析表明,Na+的电导率主要来源于8a位点的Na离子,而16d位点的Na离子主要用于形成菱形骨架。
2.3.3 氧化物电解质,
Li7La3Zr2O12(LLZO)电解质有广泛的应用,吴顺情团队为Li7La3Zr2O12 (LLZO)系统开发了深度势,将训练集和测试集的覆盖率作为训练迭代的收敛准则,其中覆盖率通过主成分分析计算。并用得到的势函数描述LLZO系统的结构和动力学特性。模拟结果准确描述了与实验相符的径向分布函数和热膨胀系数。它还预测了LLZO体系的四面体到立方相相变行为。
An Qi团队开发了LiGe2(PO4)3体系深度势,对有序和无序的LiGe2(PO4)3超胞进行采样,使其能够描述结晶、熔融和玻璃态。模拟结果显示P原子在晶态和玻璃态下均具有四面体氧环境。锗原子的配位环境更为复杂,具有晶态八面体氧环境和玻璃态混合环境,观测到4、5和6配位的Ge原子。通过取向序参数的计算表明,在玻璃态的LiGe2(PO4)3中,4配位的Ge原子具有四面体氧环境,而6配位的Ge原子具有八面体氧环境,如图12所示。
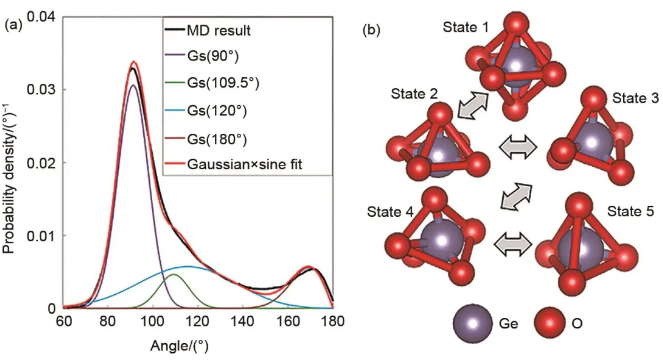
图12 (a) O-Ge-O角分布(分解为高斯函数和正弦函数的乘积);(b) 液态/玻璃态Ge原子氧环境的可能转变:1—八面体,2—方形金字塔,3—三角双金字塔,4—过渡结构,5—四面体
2.3.4 非晶电解质
非晶电解质相比于晶体电解质结构更加复杂,在采样方面要求更高,同时非晶电解质很难直接从实验上获得结构信息,深度势能模拟为其结构解析和性质计算提供了一种有效的方法。
An Qi团队构建了Li2S-SiS2-P2S5三元电解质势函数探索了不同组成的玻璃结构,在密度、弹性模量、径向分布函数和中子结构因子方面与DFT和实验结果取得了一致的结果。模拟揭示了这些玻璃相中Si和P的不同局部环境,在Li2S-SiS2中大多数Si原子处于共享边构型,而在三元Li2S-SiS2-P2S5组成中混合了共享角和共享边的四面体。300 K条件下三元玻璃的电导率为2.6 mS/cm,介于50Li2S-50SiS2(2.1 mS/cm)和75Li2S-25P2S5(3.6 mS/cm)两者之间。此外,对三元玻璃中锂离子在MD轨迹上扩散的深入分析表明,扩散路径与附近SiS4或PS4四面体的旋转动力学之间存在显著相关性。
Gerbrand Ceder团队结合深度势能模型和扩展X射线吸收精细结构谱研究了非晶2LiCl-1GaF3的行为,揭示了离子固体混合物形成软黏土的微观机制。发现阴离子交换导致的类固体分子单元的形成是软黏土类力学行为的关键。阴离子交换中形成的分子固体单元,以及这种反应的缓慢动力学,是软黏土形成的关键。扩展X射线吸收精细结构光谱证实了分子固体单元的形成。制定了从离子固体混合物中制造软黏土状材料的一般策略。在此基础上,该团队进一步开发了一种球磨法制备的电导率达到0.47 mS/cm的新型软黏土电解质xMgCl2-GaF3。通过深度势能模拟和扩展X射线谱相结合,确定了软黏土结构的形成是由剪切过程中的部分阴离子交换引起的。快速Mg离子传导归因于镁离子在富氯化学环境中的不协调。
吴顺情团队开发了一个能够准确描述具有不同元素比的非晶Li-La-Zr-O系统的原子势函数。采用了一种预训练方法,并迭代地合并了新的训练集,如图13所示。利用这个势函数,计算了不同元素比与锂离子电导率之间的关系并分析了造成影响的原因,结果表明,La-La原子之间的聚集程度比Zr-Zr原子之间的聚集程度更严重。在保持Li含量不变的同时,增加非晶结构中Zr的含量,降低La的含量,可以产生额外的空位,促进Li离子的扩散。
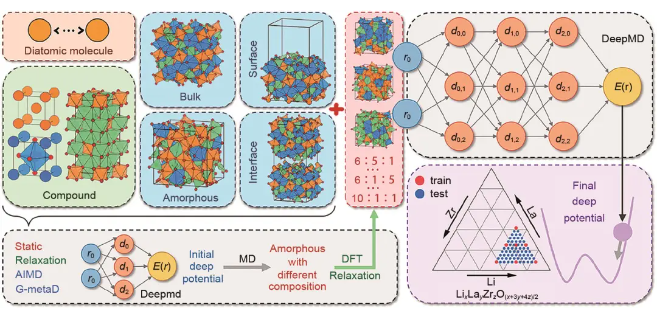
图13 非晶Li-La-Zr-O系统深度势能预训练流程
胡勇胜团队报道了一类对Li+和Na+都具有高离子电导率(30 ℃时约为1 mS/cm)的黏弹性无机玻璃电解质NaAlCl2.5O0.75和LiAlCl2.5O0.75,用DPMD研究了玻璃相的形成和离子传导机理,LiAlCl4熔体的离子在凝结过程中很容易重排成晶体,而用O原子取代部分Cl原子后,形成的Al-O-Al网络抑制了结晶而产生玻璃相。适当的O-Cl比有助于降低玻璃相的Tg从而提高离子迁移速率。
相比于对每种具体的固态电解质分别进行采样和研究,建立一个适合多种电解质的大模型对于结构搜索和结构设计更有效,深势科技团队开发的固态电解质大模型,覆盖LiXYS(X=Ge,Sn,Si; Y=P,Ga,Sb,As……)和LiPSX(X=Cl,Br,I)等40多种体系,为固态电解质的模拟提供了更多的可能。
2.4 电解液
相比于固态电解质,电解液体系有其自身的特点,固态电解质运动过程中一般以原子或几个原子形成的原子簇为基本单元,基本单元对称性高,原子局部环境空间较小;而电解液往往以分子为基本单元,结构对称性低,不同排列取向形成了更加丰富的原子局部环境空间。这使得相比于固态电解质体系,电解液体系需要更加丰富的采样,因此深度势能在电解液中的应用相比于固态电解质有一些滞后。溶液体系的结构的准确模拟本身也具有重要的意义,液态体系不同于结构周期性排布的晶体,由于涉及多种作用力的竞争且溶剂化结构处于动态变化中,传统实验方法如X射线衍射、振动光谱、核磁波谱等,都难以获得精确动态的溶剂化结构。在传统分子动力学模拟方法中,AIMD精度足够但由于计算昂贵,只能进行小的时间和空间尺度(500 atoms, 100 ps)的采样,CMD计算量小,计算速度快,可进行数万原子和约100 ns的采样,但基于经验参数的经典力场精度低。虽然其对于溶剂化结构和输运规律的基本认识起了很大作用,但当关心氧化还原性质等涉及精确溶剂化结构和电子结构的性质时,CMD便不再满足需求了。目前,深度势能已在水系电解液、有机系电解液、离子液体等液体体系中获得成功,在与AIMD对比,或与密度、扩散系数、黏度、氧化还原电位等物理化学性质实验结果的对比中展示了其可靠性。
2.4.1 水系电解液
水分子只含三个原子,离子配位空间相对简单,且深度势能已在纯水体系中有很好表现,因此水体系是最早使用深度势能建模的电解液体系。Zhang团队利用深度势能训练了锌离子水溶液的势函数,并研究了Zn2+的配位结构,DPMD模拟得到的Zn2+的镜像分布函数能很好地重现X射线吸收近边结构谱的实验结果,且计算速度相比QM或QM/MM方法快了几个数量级。陈墨涵团队使用深度势能结合杂化泛函SCAN训练了Ca2+/Mg2+离子H2O溶液的势函数,分别对Ca2+-OH-和Mg2+-OH-离子对进行1 ns的模拟,系统研究了两种阳离子对OH-溶剂化结构的影响,发现OH-对Ca2+的溶剂化结构的影响比对Mg2+的更显著。Ca2+第一溶剂化层的水分子相比Mg2+的能更快改变取向。与阳离子结合使OH-的氢键被强烈改变,导致相邻OH-的水分子被挤压。解决了之前研究中由于GGA泛函精度不足和AIMD时间尺度不足导致OH-结构和性质矛盾的问题。
DPMD不仅有助于研究电解质溶液中的溶剂化结构,还为电化学储能场景下备受关注的氧化还原电位计算提供了解决方案。氧化还原电位的准确计算需要势函数足够精确,同时对体系得电子过程的始末状态进行充分采样。深度势能提供的高精度和高速度,大大提升了这一计算的效率。程俊团队制定了合理的采样流程,进行了电解液体系的深度势能采样,并将深度势能引入了基于热力学积分方法的自由能计算工作流,相比于之前基于AIMD的方法效率大大提升。该自动化工作流为从头计算重要热力学性质如氧化还原电位和酸度提供了一种非常有效的方法,沿着反应的热力学路径更新数据集进行深度势能训练有助于保证初始状态和最终状态之间中间状态的势函数准确性,从而获得准确的自由能。该方法成功运用在LiTFSI/H2O电解液中,首次从严谨的计算电化学层面揭示了高浓度电解液提高Li金属电池循环稳定性的原因。对不同盐浓度下LiTFSI/H2O电解液中水分子以及阴离子的氧化还原电位的计算表明,随着溶液浓度的升高,离子与H2O形成的缔合网络逐渐增强,阻碍了H2O得电子形成OH-后溶剂环境的重组,降低了H2O的还原电位,导致阴离子和H2O的氧化还原电位顺序的切换,从而抑制了析氢反应的发生而有利于TFSI诱导的SEI膜的形成。后续,该组还将该方法拓展到实际溶剂化环境下的有机分子氧化还原电位和酸度的计算,并开发了一个自动化的工作流来构建始末两种状态(得失电子或质子)的通用深度势能函数,其工作流如图14所示,使性质计算更加高效低成本,为水相有机液流电池电解液的设计提供了便利。
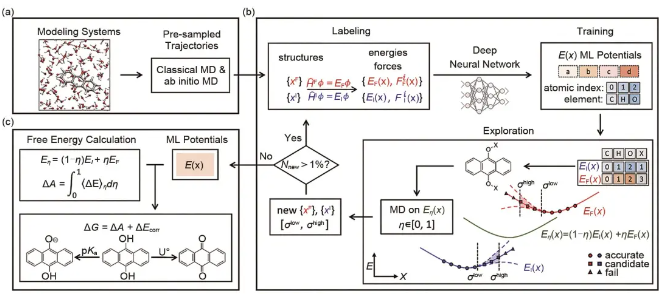
图14 酸度与氧化还原电位自动化工作流 (a) 初始化;(b) DP势函数训练过程;(c) DPMD自由能计算
另一个利用DPMD理解电解质溶液理化性质的成功案例是盐水的介电衰减,Michael L. Klein团队使用深度势能建立了NaCl-H2O模型,精确重现了实验中介电常数随浓度的下降的趋势。对模拟的深入分析表明,离子水合壳侵入溶剂氢键网络使水分子间偶极关系破坏,因此,随盐浓度升高,水溶液介电常数降低的主要原因是溶剂水的集体响应受到更大的抑制,而非过去认为的H2O分子介电响应的饱和。
2.4.2 有机系电解液
有机系电解液相比于水系电解液配位结构空间更复杂。为了在相空间中充分采样,一方面需要使用尽可能丰富的初始结构;另一方面要使用高温(500 K)进行采样,加快配位结构的变换速率。否则采样区间可能陷入初始结构附近的势能面局部最小值点。有机系电解液种类多样,但目前使用DPMD的实例还不普遍,程俊团队训练了LiTFSI/三乙二醇二甲醚(G3)的深度势能模型,系统对比了周期性体系常用泛函PBE和BLYP的表现,发现PBE-D3相比BLYP-D3拥有更好的精度。在研究配位结构时还发现,纳秒级时长的轨迹才能使Li+配位结构收敛,在更短的时间尺度内,轨迹明显受模拟初始结构影响。
赵金保团队训练了MgTFSI2/MgCl2的3-甲氧基丙胺溶液的深度势能模型,用于探究两种电解液在Mg金属电池中的不同电化学表现。对DPMD获得的精确动态溶剂结构进行进一步DFT研究,发现MgCl2盐相比MgTFSI2盐具有更快的界面电荷转移动力学是由于MgCl2更易缔合而使Mg2+离子的溶剂配体交换速率提高接近3倍。研究中还发现,400 K下二价Mg2+与溶剂分子的交换速率接近0.5 ns-1,相比于一价Li+慢了1个数量级以上,因此需要数十纳米的模拟才能对该体系的溶剂化结构进行充分采样。
徐宏团队训练了含有H3O+离子的LiPF6-EC/DMC体系的深度势能模型,用于研究H3O+对PF6-分解的影响规律。模拟结果显示极性溶剂具有铆合电解液中微量H3O+的作用,使其不易进攻PF6-从而抑制电解液分解,模拟结果与不同温度和浓度的系统性实验结果相吻合。
对于电解液体系,DPMD提供的高精度和长时间尺度模拟已在结构和宏观理化性质的预测方面发挥了重要作用,然而,立足长远看,直接模拟电解液在化学/电化学反应中的动态变化过程仍然是一个重要目标,如电解液分解、劣化等,但在采样和时间尺度方面仍有挑战。
2.4.3 离子液体
离子液体也是一类重要的电解液体系,其不含有溶剂分子,直接由阴阳离子相互间滑动实现离子传导。何宏艳团队训练了一个适用于10种室温离子液体(1-乙基-3-甲基咪唑结合不同阴离子)体系的深度势能模型。对各离子液体进行纳秒级模拟。对原子能量受力、键角分布和势能的误差进行系统分析,验证了DPMD的精确性。对振动谱和氢键的分析表明DPMD能合理预测离子间库仑相互作用和氢键相互作用的耦合性质。
DPMD在高温熔盐(高温离子液体)体系也有成功实践,唐忠锋团队训练了MgCl2-NaCl和MgCl2-KCl熔融盐的深度势能函数,并研究了其在800~1000 K温度范围内的结构和热物理性质的关系。在足够大的时间和空间尺度下,DPMD成功再现了这两种氯化物的密度、径向分布函数、配位数、平均势力、比热容、黏度和热导率。石伟群团队训练了LiCl-KCl-LiF体系的深度势能函数。并用DPMD研究了体系的局部结构、热物理性质和输运性质,并分析了温度和LiF浓度对上述性能的影响。
2.5 界面
电解质界面包含多个相,相比于单相的电解质体系更加复杂,程俊团队在惰性金属/H2O电极和半导体电极/H2O等较稳定界面的结构和性质方面做了很多工作,但电池材料中的界面更加复杂,尤其是电极材料具备反应性,其与电解质形成的界面可能存在一个动态演化过程。深度势能在负极/电解质界面、正极/电解质界面、不同电解质间的界面等方面一些探索。杨勇团队用深度势能训练了锂金属与无机固态电解质Li|β-Li3PS4界面的模型,训练需要对两相以及界面相都进行充分采样。第一步分别训练Li金属与β-Li3PS4的势函数,第二步将Li[001]面与β-Li3PS4的[100]/[010]和[001]面分别拼接,并用DP-GEN自动采样。模型训练好后进行了大空间尺度(12000原子,40 nm厚)和时间尺度(2 ns)的模拟,观察到Li|β-Li3PS4界面存在明显的演化过程,该过程可分为4个阶段,即离子快速扩散、Li2S组分成核、Li2S晶相的生长和Li2S晶相稳定化。界面形成过程中,能观察到新组分逐渐由非晶态转化为晶态,这可能揭示了界面SEI生长的一般过程。
许审镇团队用深度势能建立了正极/电解质界面,分别搭建Li6PS5Cl/LiCoO2、LiCoO2/LiF和Li6PS5Cl/LiF界面模型并再用DP-GEN主动学习,之后用于模拟LiCoO2/LiF/Li6PS5Cl三项体系,如图15所示,考察LiCoO2和Li6PS5Cl间加入非晶LiF层对界面结构演化和Li+迁移速率的影响,结果显示,当不含非晶LiF层时,Li6PS5Cl/LiCoO2将随时间演化生成P-O、Co-S、S-S等新键,表明在界面处发生了反应,而非晶LiF的存在虽不能抑制S-S键的生成,却能有效保护Li6PS5Cl中P-S四面体的局部结构,防止界面结构劣化。然而非晶LiF涂层厚度过大时,LiF相将产生有序Li-F4结构从而抑制锂离子的扩散,因此最佳非晶LiF厚度在1 nm。从两相初始接触状态直接建立模型研究界面相演化过程计算量很大,一种折中而有效的方法是,通过实验表征等方法初步确定界面相的结构,再用过DPMD研究其构效关系。许审镇团队研究了Li金属/电解液界面常会产生的Li2CO3和LiF组分,研究了在界面相中的Li+传输机制,由于500 K时,LiF会自发形成规则四面体结构,室温下锂离子在LiF中的扩散速率极低,而当这两种SEIs组分混合时,Li2CO3在抑制LiF规则四面体形成中的关键作用,使得电导率升高,并表明抑制大尺寸体相LiF的形成可能对提高电池性能至关重要。深度势能在这些方面的应用展现出其探索界面演化规律和性质的巨大潜力。
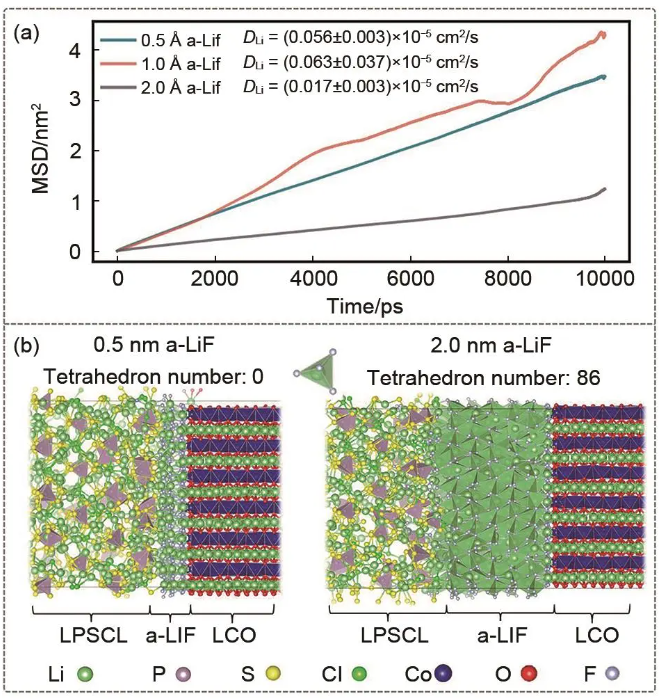
图15 (a) 不同厚度的α-LiF涂层区域内Li离子的均方位移(MSD)随模拟时间的变化及获得的扩散系数。(b) 0.5 nm和2.0 nm层厚度的LiF的原子堆积形貌。规则的Li-F四面体数和P-S四面体也被显示
3 总结与展望
深度势能模型作为一种前沿的计算材料科学工具,在电化学储能材料的研究中扮演着日益重要的角色。本文综述了DP模型在电池材料模拟中的广泛应用,包括负极材料、正极材料、固态电解质以及电解液等。通过深度学习技术,DP模型能够以较低的计算成本实现对材料体系中原子尺度行为的高精度预测,这对于理解材料的内在性质和设计新型电池材料具有重要意义。
在负极材料方面,DP模型成功应用于硅基和碳基材料,揭示了锂化/脱锂过程中的体积膨胀、结构演变和锂离子扩散机制等问题。这些发现对于开发具有高能量密度和长循环稳定性的负极材料至关重要。正极材料的研究中,DP模型在精确解析晶相结构、研究结构相变微观机理、计算Gibbs自由能以及评估迁移势垒方面展现了其独特的优势,为改善正极材料的性能提供了理论基础。
固态电解质作为全固态电池的关键组成部分,其离子传导机制的研究对于提高电池的安全性和能量密度具有重要意义。DP模型在硫化物、卤化物和氧化物固态电解质中的成功应用,为理解离子在晶体中的扩散路径和动力学提供了新的视角;电解液体系的研究中,DP模型的应用不仅提高了对溶液结构和性质的理解,还为精确物理化学性质如氧化还原电位、酸度等性质的计算提供了新的策略。
尽管DP模型在电池材料研究中取得了显著进展,但仍面临一些挑战和改进空间:
(1)DFT计算的准确性问题:DP模型的训练依赖于DFT计算提供的数据,目前DFT在处理正极材料体系需要结合Hubbard模型(DFT+U)来准确描述电子结构,在处理石墨负极材料体系仍需要结合色散校正(DFT-D)来准确描述层间结构。因此,需要进一步优化DFT方法或结合其他理论模型,以提高训练数据的准确性。
(2)长程相互作用的处理:DP模型在处理长程静电相互作用时存在不足,特别是在电解质界面等电荷分离的体系中。在模型中考虑长程静电相互作用,能更全面地描述此类体系的性质。
(3)界面反应的深入研究:电极/电解质界面是电池材料中的关键区域,其动态演化过程对电池性能有直接影响。DP模型需要进一步发展,以更准确地模拟界面反应和离子传输机制。
(4)多尺度模拟的结合:DP模型可以与量子力学、分子动力学和连续介质模型等多尺度模拟方法相结合,以全面理解材料从原子到宏观尺度的行为。
深度势能模型作为一种强大的计算工具,在电化学储能材料的研究中展现出巨大的应用潜力。通过不断的模型优化和算法创新,DP模型有望在未来的材料设计和电池技术发展中发挥更加关键的作用。同时,DP模型的发展也需要与实验研究紧密结合,以实现对材料性质更深入和全面的理解。随着计算资源的日益丰富和算法的不断进步,有理由相信,DP模型将在推动新能源材料的发展和电池技术革新中扮演更加重要的角色。

评论